
تحليل متعمق لإعداد الملفات الفنية وآليات الموافقة على تسويق وطرح المستحضرات الدوائية في مختلف انحاء العالم
استكشف مقالنا الشامل حول ملف المستحضر وتراخيص التسويق، حيث يُلقي الضوء على مفهوم الإضبارة ودورها في تقييم المنتجات الدوائية. نقدم لك نظرة مفصلة على تراخيص التسويق وكيفية استعراض ملفات التسجيل لتحقيق موافقة على تسويق الدواء على مستوى عالمي. اكتشف كيف تقوم هيئات التنظيم المختلفة بتقسيم العمليات والتحقق من الامتثال باستمرار.
الإضبارة (Dossier) هي مجموعة من الوثائق حول مواضيع محددة. تخضع أي عملية تحضير لمنتج دوائي معد للاستخدام البشري لعملية استعراض وتقييم ملف المنتج (الاضبارة) الذي يحتوي على معلومات تفصيلية حول الجوانب الإدارية والجودة والبيانات غير السريرية والسريرية. تصان او تقيم هذه العملية بواسطة سلطة التنظيم الدوائية للبلد وتُعرف هذه العملية باسم NDA في الولايات المتحدة وMAA في الاتحاد الأوروبي وفي البلدان الأخرى باسم Registration Dossier. تقسم هذه العملية الى نوعين وهما ICH-CTD وACTD. يتبع ICH-CTD من قبل دول ICH وكذلك الدول العالم الثالث أو النامية، بينما يتبع ACTD من قبل جمعية دول جنوب شرق آسيا (ASEAN). يعتبر ACTD جسراً بين متطلبات التنظيم في الدول المتقدمة والنامية. إذا حدث توافق بين الإرشادات الخاصة بـ CTD و ACTD، يؤدي ذلك الى تقليل الفجوة والاختلاف والتباين بين الإرشادين.
يعرف ترخيص التسويق أو التسجيل أو الترخيص بأنه عملية نقد وتقييم إضبارة المستحضر التي تحوي على تفاصيله الإدارية والكيميائية والسريرية وغير السريرية والإذن الممنوح من قبل الجهات التنظيمية في البلد بهدف دعم تسويقه أو الموافقة على طرحه.
ملف التسجيل لمنتج الأدوية هي وثيقة تحتوي على جميع البيانات التقنية (الإدارية ومعايير الجودة والتفاصيل غير السريرية والسريرية) لمنتج دوائي يُعتمد/يُسجل/يُسوق في بلد معين. وتُعرف عادة باسمNew Drug Application (NDA) في الولايات المتحدة أو Marketing Authorization Application (MAA) في الاتحاد الأوروبي وفي الدول الأخرى باسمRegistration Dossier.
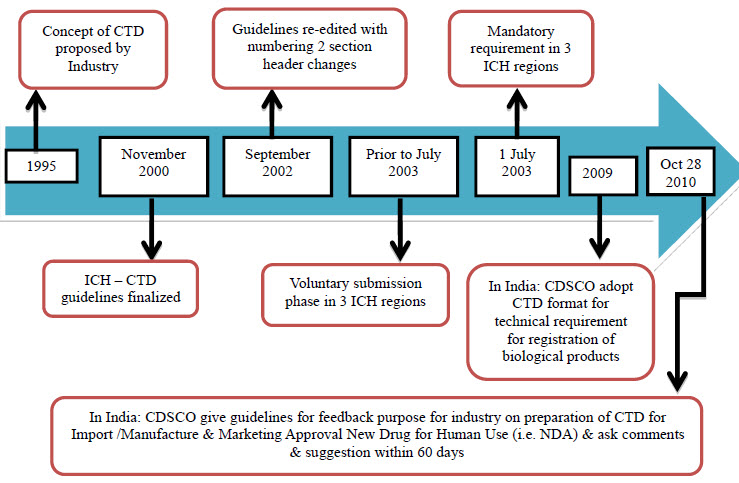
قام مؤتمر التنسيق الدولي للمتطلبات الفنية الخاصة بتسجيل الأدوية المعدة للاستخدام البشري (ICH-The International Conference on Harmonization) بتوحيد ترتيب وثائق التسجيل من خلال إصدار إرشاد الملف الفني المشترك (CTD- Common Technical Document). هذا التنسيق الموصى به في إرشاد CTD لتقديم طلبات التسجيل أصبح مقبولًا على نطاق واسع من قبل السلطات التنظيمية داخل مناطق ICH وخارجها.
الاضبارة هي ملف يجب تقديمه استنادًا إلى متطلبات موافقة الدواء/ترخيص التسويق، فهي وثيقة علمية شاملة تُستخدم للحصول على موافقة ترخيص التسويق العالمي للدواء من قبل سلطات الصحة المتنوعة. يعتمد إعدادها ومعالجتها وتجميعها وإرسالها ميدانياً من قبل إدارة شؤون التنظيم على العديد من الأنشطة المترابطة، وسيتم الاعتماد على المنطقة التي سيتم طرح الدواء فيها في وضع اليات عملية التعبئة والتفويض في السوق.
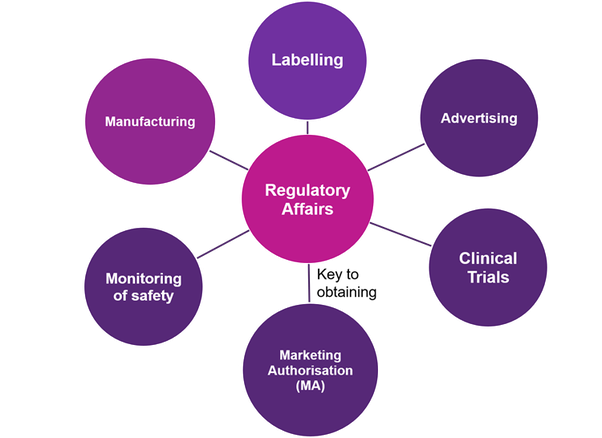
تطوير صناعة الأدوية على مستوى عالمي أدى إلى الحاجة إلى توحيد التوصيات لتطوير الأدوية الجديدة، فضلاً عن متطلبات التنظيم المختلفة في البلدان المختلفة. وبالتالي، سيساعد تبني التنسيق المشترك في التقديم على التغلب على هذه العقبات. من خلال عملية ICH، تم تطوير إرشادات CTD لليابان والاتحاد الأوروبي والولايات المتحدة. تقريباً معظم الدول قد اعتمدت تنسيق CTD.
المستند الفني المشترك (CTD)
CTD هو مجموعة من المواصفات لملف الطرح لتسجيل الأدوية وتصميمها للاستخدام في جميع أنحاء أوروبا واليابان والولايات المتحدة. تم تطويره بواسطة وكالة الأدوية الأوروبية (EMA) ووكالة الغذاء والدواء (FDA) ووزارة الصحة والعمل والرفاه (اليابان). يتم الحفاظ على CTD بواسطة المؤتمر الدولي لتنسيق المتطلبات التقنية لتسجيل الأدوية المعدة للاستخدام البشري. لقد غيَّر هذا التنسيق من الية تجميع المعلومات المتعلقة بالجودة والسلامة والفعالية في إطار مشترك لعمليات مراجعة التنظيم.
اعتبارات عامة
– CTD هو تنسيق موحد لتقديم المعلومات إلى السلطات التنظيمية ذات الصلة. فهو نموذج لتقديم البيانات في الملف. و يشير إلى تنسيق مناسب للبيانات التي تم الحصول عليها.
– CTD ليس تصريح معلن للبيانات. وليس إرشاد يهدف إلى تحديد الدراسات المطلوبة او تحديد المحتوى.
يشترط ما يلي:
- يحتوي على معلومات واضحة ودقيقة
- ذو نمط وحجم خط محدد سهل القراءة
- يتبع إرشادات ICH للترقيم والتفصيل التوثيقي
- يحتوي على جميع الاختصارات المستخدمة وتكون مدرجة في نهاية الملف.
- يقدم معلومات صحيحة حول المواد الأولية الداخلة في تصنيع التركيبة النهائية.
هيئات التنظيم للـ CTD
1) التنظيم بموجب قانون الأدوية ومرسوم 122A و122B و122D والملحق الأول والأول A والسادس من الجدول Y يصف المعلومات المطلوبة للموافقة على طلب استيراد أو تصنيع دواء جديد للتسويق.
2) كل بلد لديه سلطة تنظيمية خاصة به مسؤولة عن فرض القواعد واللوائح وإصدار إرشادات لتطوير الدواء والترخيص والتسجيل والتصنيع والتسويق وعنونة المنتجات الدوائية.
3) جميع الدول المستقلة في العالم لديها سلطات تنظيمية خاصة بها.
تم توقيع CTD رسمياً في نوفمبر 2000، في الذكرى العاشرة للمؤتمر الدولي للتنسيق؛ سان دييغو، كاليفورنيا.
منظمة الصحة العالمية (WHO)
عندما اجتمع الدبلوماسيون لتأسيس الأمم المتحدة في عام 1945، كانت إحدى الأمور التي ناقشوها إنشاء منظمة صحية عالمية.
دستور منظمة الصحة العالمية: تم اعتماد الدستور في المؤتمر الصحي الدولي الذي عقد في نيويورك من 19 يونيو إلى 22 يوليو 1946 وتم توقيعه في 22 يوليو 1946 من قبل ممثلين عن 61 دولة ودخل حيز التنفيذ في 7 إبريل 1948 – وهو تاريخ نحتفل به الآن سنويًا باسم “يوم الصحة العالمي”.
منظمة الصحة العالمية البان أمريكية (PAHO-Pan American Health Organization)
تأسست منظمة الصحة العالمية البان الأمريكية (PAHO) في عام 1902، وهي أقدم وكالة دولية للصحة العامة في العالم. تقدم التعاون التقني والتنسيق بين الشراكات لتحسين الصحة وجودة الحياة في بلدان الأمريكتين. PAHO هي وكالة الصحة المتخصصة في نظام البان الأمريكي وتعمل كالمكتب الإقليمي لأمريكا في منظمة الصحة العالمية (WHO). إلى جانب WHO، تعتبر PAHO عضوًا في نظام هيئة الأمم المتحدة.
منظمة التجارة العالمية (WTO-World Trade Organization)
هي منظمة قائمة على القوانين وتعتمد على الأعضاء – جميع القرارات تتخذ من قبل حكومات الأعضاء والقوانين هي نتيجة التفاوض بين الأعضاء.
تتمثل مهمة ICH في تقديم توصيات لتحقيق توحيد أكبر في تفسير وتطبيق الإرشادات والمتطلبات الفنية لتسجيل المنتجات الصيدلانية، مما يقلل أو يلغي تكرار الاختبارات التي تجري خلال بحث وتطوير الأدوية البشرية الجديدة.
تأسست ICH في عام 1990 كمبادرة فريدة تجمع بين سلطات تنظيم الأدوية وصناعة الأدوية في أوروبا واليابان والولايات المتحدة.
تقدم توحيد التنظيم العديد من الفوائد المباشرة لكل من سلطات التنظيم وصناعة الأدوية بتأثير إيجابي على حماية الصحة العامة.
التقدم في التنسيق الدولي
حدثت تغييرات هائلة في المتطلبات الفنية والعلمية للملف، حيث استمرت أعمال ICH في وضع واعتماد إرشادات جديدة في فئات الجودة والسلامة والفعالية والتخصصات المتعددة. هيكل CTD الموحد والتنسيق الوحيد لملفات تسجيل المنتجات الدوائية الجديدة التي تم اعتمادها في سان دييغو، هو الآن التنسيق الإلزامي في الاتحاد الأوروبي (EU) واليابان وكندا وسويسرا وأستراليا. وهو التنسيق الموصى به في الولايات المتحدة.
بذلت جهود ضخمة من قبل موظفي الوكالات التنظيمية وصناعة الأدوية في أعمال ICH، وقد تحققت درجة عالية من التنسيق في العديد من المجالات العلمية والتقنية للملف. وعلى الرغم من ذلك، لا تزال هناك فروق وطنية في محتوى التقديمات، ليس فقط في الوحدة 1 والمعلومات الإدارية والوصفية، ولكن أيضًا في مجالات أخرى من الملف. وتنشأ هذه الفروق من الاختلافات في الممارسات والإجراءات التنظيمية، وممارسات الطب والصيدلة المختلفة والفروق في الوصول إلى الإجراءات التشخيصية والعلاجية. ومع ذلك، نحن لا نزال بعيدين عن الحصول على ملف تسجيل وحيد على مستوى عالمي بمعنى حقيقي.
زيادة التعاون بين الوكالات بناءً على ICH
اتفاقيات الاعتراف المتبادل بشأن ممارسة التصنيع الجيدة (GMP) قائمة بين الاتحاد الأوروبي وكندا وأستراليا ونيوزيلندا وسويسرا واليابان. توجد اتفاقيات بين العديد من البلدان (بما في ذلك أعضاء ICH) لتبادل معلومات مراقبة الأدوية ومعلومات عيوب المنتج.
تتيح اتفاقيات السرية بين الاتحاد الأوروبي وإدارة الأغذية والأدوية (FDA) الآن تبادل المعلومات حول القضايا القانونية والتنظيمية والمشورة العلمية وتسمية الأدوية اليتيمة وتقارير الفحص وإجراءات الترخيص ورصد السوق بعد التسويق. في سبتمبر 2004، أطلقت الوكالة الأوروبية للأدوية (EMEA) وإدارة الأغذية والأدوية (FDA) برنامجًا تجريبيًا لاجتماعات المشورة العلمية المتوازية للرعاة للحصول على مشورة بشأن القضايا العلمية خلال مرحلة تطوير المنتجات الدوائية الجديدة. تم استهداف المنتجات اليتيمة (الأدوية التي تستخدم لعلاج الأمراض النادرة) والمنتجات الخاصة بالأطفال بشكل خاص. زاد التعاون الثنائي والثلاثي في عام 2008. وافقت هيئة الصحة الكندية على تبادل المعلومات مع اللجنة الأوروبية وEMEA حول ترخيص وسلامة الأدوية. بدأت كندا وأستراليا مشروعهما لاستعراض الموازي للمنتجات البيولوجية (الذي أُطلق في الأصل في عام 2006). وكل هذا لم يكن ممكنًا إلا استنادًا إلى العمل السابق الذي تم في توحيد متطلبات التنظيم وفي تطوير تنسيق ملف التسجيل في ICH.
مبادرات التنسيق الإقليمي
تم إنشاء عدة مبادرات لتحقيق التنسيق الإقليمي حيث يقوم تجمع جغرافي من الدول بتنسيق المتطلبات الفنية والعلمية، وفي بعض الحالات، تنسيق شكل ملف التقديم للدول الأعضاء. تم دعوة هذه الفئات لتعيين ممثلين دائمين للجنة العامة للتنسيق. حالياً، تشمل هذه المجموعات:
- منتدى التعاون الاقتصادي لدول آسيا والمحيط الهادئ (APEC): يحوي 21 دولة في منطقة آسيا والمحيط الهادئ.
- رابطة دول جنوب شرق آسيا: بروناي ودار السلام، كمبوديا وإندونيسيا ولاوس وماليزيا وميانمار والفلبين وسنغافورة وتايلاند وفيتنام.
- مجلس التعاون الخليجي: المملكة العربية السعودية الكويت والإمارات العربية المتحدة وعمان والبحرين وقطر واليمن.
- شبكة بان الامريكية لتحقيق التنسيق في مجال تنظيم الأدوية (PANDRH): الأرجنتين وباربادوس وبوليفيا والبرازيل وتشيلي وكولومبيا وكوستاريكا وكوبا وجواتيمالا وجامايكا والمكسيك وبنما وترينيداد وتوباغو وفنزويلا.
- مجتمع تنمية جنوب إفريقيا (SADC): أنغولا وبوتسوانا وجمهورية الكونغو الديمقراطية وليسوتو ومالاوي وموريشيوس ومدغشقر وناميبيا وسيشل وجنوب أفريقيا وسوازيلاند وتنزانيا وزامبيا وزيمبابوي.
تقوم هذه المجموعات الإقليمية بمراجعة تطبيق إرشادات المؤتمر الدولي لتنسيق متطلبات التجارب السريرية (ICH) في بلدانها الخاصة. وتشمل المواضيع ذات الاهتمام الخاص إرشادات استقرار تنسيق متطلبات الجودة (ICH)، وإرشادات الممارسات الجيدة للتصنيع (GMP)، ومتطلبات دراسات التوافر والتكافؤ الحيوي والتجارب السريرية وتصدير/استيراد الأدوية والأدوية التقليدية ورصد السوق.
المناطق التي تتبع نهج أو تنسيق ال CTD الخاص بتسجيل المنتج:
* منطقة آسيان ASEAN Region (تنسيق ACTD): بروناي وكمبوديا وإندونيسيا وجمهورية لاو الديمقراطية وماليزيا وميانمار والفلبين وسنغافورة وتايلاند وفيتنام.
* المنطقة الإفريقية (تنسيق CTD): نيجيريا وكينيا وجنوب أفريقيا وزيمبابوي وتنزانيا وإثيوبيا وناميبيا ووموريشيوس.
* المنطقة السيس CIS Region (تنسيق CTD) (يشبه تنسيق CTD لكل بلد): مولدوفا وروسيا وأوكرانيا وجورجيا وكازاخستان وأوزبكستان.
* منظمة الصحة العالمية والهند (تنسيق CTD)
* المنطقة اللاتينية (تنسيق محدد لكل بلد): المكسيك وبنما وفنزويلا وتشيلي وكوستاريكا والبرازيل وجمهورية الدومينيكان وجامايكا.
* منطقة مجلس التعاون الخليجي (تنسيق CTD) وأستراليا (تنسيق ICH-CTD). إجراءات التسجيل تختلف من منطقة إلى أخرى. وبالتالي، يتبع بعضها إرشادات المؤتمر الدولي لتنسيق متطلبات التجارب السريرية (ICH)، وتتبع بعضها إرشادات منظمة الصحة العالمية (WHO) لتسجيل منتج الدواء. ولكن بعض المناطق لديها إرشادات خاصة بالبلد لتسجيل المنتج الصيدلاني النهائي (FPP). في صناعات الأدوية، تفرض شؤون التنظيم الدوائي ملفين هما CTD (الملف الفني المشترك) و ACTD (الملف الفني المشترك لدول جنوب شرق آسيا).
تنظيم تنسيق ICH-CTD:
تنظم الـ CTD إلى خمسة وحدات. الوحدة 1 محددة للمنطقة. الوحدات 2 و 3 و 4 و 5 تكون مشتركة لجميع المناطق. ينبغي أن يكون الامتثال لإرشادات المؤتمر الدولي لتنسيق متطلبات التجارب السريرية (ICH) يضمن تقديم هذه الوحدات الأربع بتنسيق مقبول لدى منظمة الصحة العالمية والسلطات التنظيمية.
الوحدات الخمس هي:
– الوحدة 1: المعلومات الإدارية والتوجيهية
– الوحدة 2: نظرة عامة وملخص للوحدات 3 إلى 5
– الوحدة 3: الجودة (التوثيق الصيدلاني)
– الوحدة 4: السلامة (دراسات التسمم/غير السريرية)
– الوحدة 5: الفعالية (الدراسات السريرية)
يظهر الرسم البياني أدناه هذه المعلومات كهيكل قائم على الوحدات المعروف باسم مثلث CTD.
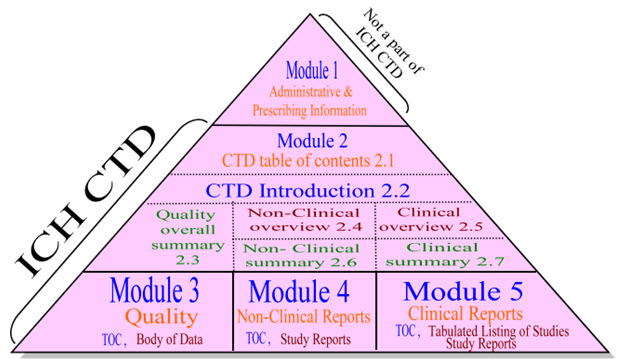
يظهر أن الوحدة 1 ليست جزءًا من الـ CTD، بل تحتوي فقط على المعلومات الإقليمية أو المعلومات الإدارية للشخص الذي لديه الحق في تقديم الملف للحصول على ترخيص السوق.
تُشرح هذه الصيغة بأمثلة من بلدين مختارين هما زيمبابوي وأستراليا من قارات مختلفة يتبع كل منهما تنسيق ICH-CTD لإعداد الملف لتسجيل الدواء المختار، وهما Regorafenib و Roflumilast على التوالي.
فئات المنتجات الصيدلانية التي يمكن تقديمها كملف تسجيل CTD
يمكن تقديم معظم فئات المنتجات كملف تسجيل CTD في جميع المناطق التابعة للمؤتمر الدولي لتنسيق متطلبات التجارب السريرية (ICH). وتشمل هذه:
– منتجات مادة الدواء الجديدة (NCE)
– منتجات الأدوية الحيوية/البيوفارما
– الأشعة الصيدلانية
– الأدوية النباتية (الأدوية العشبية)
في حالة الولايات المتحدة، يمكن أيضًا تقديم ملفات تسجيل CTD للمنتجات العامة والمباعة بدون وصفة طبية وهذا التنسيق سيكون إلزاميًا في الاتحاد الأوروبي لهذه المنتجات.
اختلافات في محتوى CTD بين مناطق المؤتمر الدولي لتنسيق متطلبات التجارب السريرية (ICH)
كما ذكر في المقدمة، يعد CTD تنسيقًا موحدًا لملفات التسجيل. ومع ذلك، لا يزال المحتوى غير موحد تمامًا. يوجد العديد من الاختلافات الوطنية أو الإقليمية في محتوى التقديم، ليس فقط في الوحدة 1 ولكن أيضًا في أجزاء أخرى من الملف. وتنشأ هذه الاختلافات من الاختلافات في الممارسات التنظيمية والإجراءات التنظيمية، واختلافات في ممارسات الطب والصيدلة، واختلافات في الوصول إلى الإجراءات التشخيصية والعلاجية.
اختلافات في الوحدة 1
على الرغم من أن CTD يشير إلى الوحدة 1 باعتبارها “معلومات إدارية إقليمية” (مثل نموذج الطلب والتسمية ونص الوصف الطبي ومعلومات المريض)، إلا أن هناك اختلافات أخرى في الواقع وعادةً ما تقوم الشركات بإعداد ورقة البيانات الأساسية المشتركة لتقديمها للممارسين الصحيين لاستخدامها لإعداد نص الوصف الطبي ومعلومات المريض في الولايات المتحدة، وملخص خصائص المنتج (SmPC)، ونشرة معلومات المريض في الاتحاد الأوروبي.
الاختلافات الجوهرية في دول الاتحاد الأوربي:
- معلومات حول الخبراء (الذين يوقعون ملخصات الوحدة 2)
- متطلبات محددة لأنواع مختلفة من التقديمات (ملخصات لدعم الأدوية والتطبيقات الهجينة والتطبيقات الببليوجرافية(الاصطلاحات واللغويات غير العربية) وتطبيقات التمتع بحقوق السوق الموسعة وتراخيص التسويق الشرطية، إلخ.)
- تقييم المخاطر البيئية
- معلومات تتعلق بامتياز السوق الخاص بالأدوية اليتيمة
- معلومات تتعلق بمراقبة السلامة الدوائية والتيقظ الدوائي
- معلومات تتعلق بالتجارب السريرية
الاختلافات الجوهرية في الولايات المتحدة الامريكية:
- معلومات براءات الاختراع والحقوق الحصرية
- معلومات إدارية حول الأطفال
- خطط إدارة المخاطر
الاختلافات الجوهرية في اليابان
- حالة براءة الاختراع
- خلفية المنشأ والاكتشاف والتطوير
- قائمة بالمنتجات ذات الصلة
- بيانات لاستعراض التعيين كمواد سامة، مواد ضارة، إلخ.
- مسودة البروتوكول الأساسي لمراقبة السوق بعد التسويق
الاختلافات الجوهرية في استراليا
- معلومات حول الخبراء (الذين يوقعون على ملخصات الوحدة 2)
- متطلبات محددة حول أنواع مختلفة من التقديمات (استناداً إلى الدراسات المرجعية ومنتجات الأدوية اليتيمة والكائنات المعدلة وراثياً والأدوية المسوقة بشكل مشترك، إلخ.)
- ملفات سجل الدواء والبلازما وشهادات مطابقة النسخة الأوروبية للأدوية
- رسائل تصريح GMP (ممارسات تصنيع الأدوية الجيدة)
- ملخصات الدراسات الحيوية
- المخاطر البيئية للأدوية التي لا تحتوي على كائنات معدلة وراثياً
- بيانات مقاومة الصادات الحيوية
اختلافات في الوحدة 2
على الرغم من أن متطلبات المؤتمر الدولي لتنسيق متطلبات التجارب السريرية (ICH) الرسمية لملخصات الوحدة 2 متطابقة في جميع الدول الأعضاء في ICH وغيرها، لكن هناك احتمال وجود اختلافات إقليمية خاصة فيما يتعلق بمحتوى الوحدة 3: الجودة والوحدة 5: الدراسات السريرية في الملفات المتكاملة وسيتم عكس هذه الاختلافات في هذه الملخصات على المستوى العالي. على سبيل المثال، عندما يكون دواء الفارماكوبيوبيا موضوعًا لملف تصريح الدواء (DMF)، سيكون هناك مرجع فقط إلى DMF في الوحدة 2 من ملف تقديم في الولايات المتحدة أو اليابان، في حين سيكون هناك ملخص للجزء المفتوح من ملف الإتقان الفعّال للمادة الفعّالة في الاتحاد الأوروبي (DMF).
اختلافات في الوحدة 3
تتواجد اختلافات إقليمية. يُستشهد في ملف التنسيق الدولي (CTD) بأمثلة على ذلك كما يلي:
– سجلات الوجبات الدوائية التنفيذية لمادة الدواء والمنتج الدوائي (الولايات المتحدة فقط): نسخ من السجلات مع تحديد المعدات وظروف التصنيع وسجلات التعبئة ومعلومات التوازن (العائد النظري والعائد الفعلي والعائد المعبأ).
– عمليات التحقق من طرائق التصنيع لمادة الدواء والمنتج الدوائي (الولايات المتحدة فقط)
– بروتوكولات المقارنة (الولايات المتحدة فقط)
– خطة التحقق من العملية (الاتحاد الأوروبي فقط)، بما في ذلك بروتوكول التحقق من العملية حيث لا تكتمل دراسات التحقق من العملية التصنيعية للمنتج الدوائي
– الجهاز الطبي الذي يستخدم بالتزامن مع المنتج الدوائي (الاتحاد الأوروبي فقط)
بالإضافة إلى ذلك، قد يكون هناك حاجة إلى توفير وثائق مختصرة حول تصنيع مادة الدواء في وصف لعملية التصنيع وفي وصف لعملية التصنيع ومراقبة العملية للمنتج الدوائي في البلدان التي لا تحترم دائمًا سرية البيانات.
قد تتواجد أيضًا اختلافات في احتياجات التسويق للبلدان المختلفة. وبالتالي، عادةً يتم اعتماد مواد البلاستيك عالية الكثافة (HDPE) للأقراص أو الكبسولات في الاتحاد الأوروبي بينما في الولايات المتحدة يُستخدم زجاج البولي إيثيلين عالي الكثافة (HDPE) بشكل أكثر شيوعًا. وسيتم شرح الاختلافات الخاصة بالجودة والتركيب والسلامة في المقال القادم
تتطلب صناعة الأدوية توحيدًا وتنسيقًا دوليًا للتغلب على التحديات المتعددة. من خلال التفاعل بين الإضبارة وتراخيص التسويق، يمكن أن يكون لتنسيق CTD تأثير كبير على تسهيل عمليات المراجعة وتجاوز الاختلافات القانونية. يجسد هذا المقال دعمًا لتحسين الجودة والسلامة والفعالية في مجال تسويق المنتجات الدوائية، مع الالتفات إلى الأنظمة التي توجه هذا الجهد، بما في ذلك المؤتمر الدولي لتنسيق المتطلبات التقنية لتسجيل الأدوية المعدة للاستخدام البشري (ICH)
المرجع: