
تحليل متعمق لإعداد الملفات الفنية وآليات الموافقة على تسويق وطرح المستحضرات الدوائية في مختلف انحاء العالم -الجزء الثاني
استكشف الجزء الثاني من مقالنا حول ملف المستحضر وتراخيص التسويق، حيث يُلقي الضوء على مفهوم الإضبارة ودورها في تقييم المنتجات الدوائية. نقدم لك نظرة مفصلة على تراخيص التسويق وكيفية استعراض ملفات التسجيل لتحقيق موافقة على تسويق الدواء على مستوى عالمي.
يعد CTD تنسيقًا موحدًا لملفات التسجيل. ومع ذلك، لا يزال المحتوى غير موحد تمامًا. يوجد العديد من الاختلافات الوطنية أو الإقليمية في محتوى التقديم، ليس فقط في الوحدة 1 ولكن أيضًا في أجزاء أخرى من الملف. وتنشأ هذه الاختلافات من الاختلافات في الممارسات التنظيمية والإجراءات التنظيمية، واختلافات في ممارسات الطب والصيدلة، واختلافات في الوصول إلى الإجراءات التشخيصية والعلاجية.
الاختلافات الجوهرية في الوحدة 3
الاتحاد الأوربي:
- يمكن تقديم بيانات المادة الدوائية كجزء من ملف تصريح الصنع كجزء من نص الاتحاد الأوروبي المكون من جزئين أو كإشارة إلى شهادة ملاءمة للدستور الأوروبي.
- الثبات: يجب تحديد متطلبات التخزين وفقًا لإرشادات لجنة الأدوية البشرية الأوروبية (CHMP).
- الوصف والتركيب: يجب أن تكون الألوان مدرجة في قائمة الألوان المسموح بها في الاتحاد الأوروبي. يجب تعيين السواغات بحيث تتفق مع دستور الأدوية الأوروبية حيث يوجد نص مخصص لها.
الولايات المتحدة الأمريكية:
- يمكن الرجوع في الملف إلى المعلومات المقدمة في ملف تصريح الصنع (DMF) المزودة مباشرة من قبل مصنع المادة الدوائية إلى إدارة الأغذية والأدوية الأمريكية (FDA).
- الثبات: يجب أن تُحدد متطلبات التخزين وفقًا لمتطلبات ال FDA.
- السواغات: يجب أن تتفق مع الدستور الأوروبي إذا تم وصفها في نص مخصص.
- رقابة المنتج الدوائي: يجب أن يكون حدود التحليل ضمن ± 5% ما لم يكن هناك مبرر للتغيير. قد يكون هناك حاجة إلى مواصفات تصنيع وفترة صلاحية مختلفة. يجب أن تتفق المنتجات مع المواصفات العامة للدستور الاوربي.
- نظام إغلاق الحاوية: لا يلزم ذكر اسم الشركة المصنعة ما لم يكن المنتج حرجًا (على سبيل المثال، المستحضرات المعدة للحقن).
- الثبات: يجب أن تكون متطلبات التخزين وفقًا لإرشادات لجنة الأدوية البشرية الأوروبية (CHMP).
- يمكن الرجوع في الملف إلى معلومات ملف تصريح الصنع المقدمة مباشرة إلى إدارة الأغذية والأدوية (FDA) من قبل مصنعي السواغات وعبوات التعبئة/إغلاق العبوة.
- الوصف والتركيب: يجب أن تكون الألوان مدرجة في قائمة الألوان المسموح بها من قبل إدارة الأغذية والدواء الأمريكية (FDA). يجب تعيين السواغات بأنها تتفق مع ال(USP/NF) حيث يوجد نص مخصص.
- السواغات: يجب أن تتفق مع الدستور الأمريكي (USP/NF) إذا تم وصفها في نص مخصص.
- رقابة المنتج الدوائي: يُسمح بحدود التحليل بحيث تصل إلى ± 10%. يُسمح بمواصفة تنظيمية واحدة (فترة صلاحية).
- نظام إغلاق الحاوية: يُطلب ذكر اسم الشركة المصنعة.
اليابان:
- يمكن الرجوع في الملف إلى المعلومات المقدمة في ملف التصريح الصنع (DMF) المزودة مباشرة من قبل مصنع مادة الدواء.
- الوصف والتركيب: يجب أن تكون الألوان مدرجة في القائمة المسموح بها في اليابان.
- السواغات: يجب أن تتفق مع النصوص التنظيمية اليابانية أو السواغات الصيدلانية اليابانية.
كندا:
- يمكن الإشارة في الملف إلى المعلومات المقدمة مباشرة في ملف التصريح بالصنع (DMF) من قبل مصنع مادة الدواء.
- الحاوية/الإغلاق: يمكن الرجوع إلى ملف تصريح الصنع من المورد.
- الاستقرار: يجب أن تكون شروط التخزين مطابقة لمتطلبات وكالة الصحة الكندية (على سبيل المثال، التخزين عند درجة حرارة الغرفة المضبوطة).
استراليا:
- يمكن تقديم بيانات المادة الدوائية كجزء من ملف تصريح الصنع المؤلف من جزئين أو كإشارة إلى شهادة ملاءمة الدستور الأوربي.
- الوصف والتركيب: يجب أن تكون الألوان مدرجة في القائمة المسموح بها في أستراليا للألوان المستخدمة في المنتجات الفموية.
- الثبات: يجب أن تشير شروط التخزين إلى قائمة الإدارة الأسترالية للأدوية والأجهزة الطبية (TGA) لشروط التخزين المقبولة (على سبيل المثال، تخزين أقل من 30 درجة مئوية)
الوحدة 4:
لا يوجد فروقات جوهرية في هذه الوحدة
الوحدة 5:
الاختلافات الجوهرية في الوحدة
دول الاتحاد الأوربي:
- عند الضرورة، يجب أن تستخدم دراسات التكافؤ للمنتجات العامة وجبة أوروبية من المنتج المرجعي.
- يجب أن تتوافق التجارب السريرية عادةً مع إرشادات فعالية لجنة الأدوية البشرية الأوروبية (CHMP) حيثما تكون متاحة.
- تعتبر التجارب السريرية لمنتجات الأدوية الجديدة مقابل “المعالجة القياسية الذهبية” المصرح بها في أوروبا هامة، بالإضافة إلى دراسات البلاسيبو.
الولايات المتحدة الأمريكية:
- عادةً ما يجب أن تتوافق التجارب السريرية مع إرشادات التنظيم التي تصدرها إدارة الأغذية والدواء الأمريكية (FDA) حيثما تكون متاحة.
- يجب تضمين ملخصات السلامة والفعالية المتكاملة (ISS/ISE) الخاصة بإدارة الأغذية والدواء الأمريكية (FDA) في تقارير التحليل من أكثر من دراسة
اليابان:
- قد يكون من الضروري إجراء دراسات “الجسر” الدوائية والسريرية للسماح بتحويل البيانات الأجنبية لتكون قابلة للاستخدام في السكان اليابانيين إذا كانت الدراسات السريرية قد تمت خارج اليابان.
إدارة الاختلافات
إذا كانت الشركات ترغب في تقديم ملف في الأسواق الرئيسية في العديد من البلدان المتقدمة في العالم، يجب تصميم برنامج التطوير الكيميائي والصيدلاني والسريري وغير السريري لتلبية جميع احتياجات التنظيم الفردية لكل سوق. على سبيل المثال، قد تكون هناك حاجة إلى إجراء دراسات إضافية في العلوم غير السريرية أو السريرية كجزء من جهود “الجسر” لدعم ملف تسجيل أجنبي في اليابان. يمكن أن تكون معظم الوثائق في الوحدات من 2 إلى 5 لملف تسجيل الدواء الجديد الرئيسي متطابقة، ولكن في حال وجود اختلافات وطنية في المتطلبات (على سبيل المثال، اختلافات في 3.2.P.5.1 مواصفات المنتج الدوائي فيما يتعلق بحدود التحليل للاتحاد الأوروبي والأسواق الأمريكية)، يكون من المفضل عادةً إعداد نسختين من الوثيقة في نفس الوقت. يمكن إعداد Module 2.3 Quality Overall Summary وModule 2.5 Clinical Overview كوثائق أساسية متطابقة، ولكن يجب أن يتم مراجعتهما بواسطة الموظفين في البلد المعني وتخصيصهما حسب الحاجة لتلبية أي متطلبات تقنية أو تنظيمية مختلفة للوكالات المختلفة.
اعتماد تنسيق CTD خارج منطقة ICH
يتم اعتماد تنسيق CTD بتعديلات محلية حسب الحاجة من قبل وكالات التنظيم الوطنية الأخرى وتجمعات الوكالات الإقليمية. تضم رابطة دول جنوب شرق آسيا (ASEAN) بروناي ودار السلام وكمبوديا وإندونيسيا ولاوس وماليزيا وميانمار والفلبين وسنغافورة وتايلاند وفيتنام.
جمعية دول جنوب شرق آسيا (ASEAN)
جمعية دول جنوب شرق آسيا (ASEAN) هي منظمة إقليمية تتألف من عشر دول أعضاء، وهي: بروناي دار السلام وكمبوديا وإندونيسيا ولاوس وميانمار، ماليزيا، الفلبين، سنغافورة، تايلاند، وفيتنام.
تأسست ASEAN في 8 أغسطس 1967 على يد حكومات خمس دول – إندونيسيا وماليزيا والفلبين وسنغافورةةوتايلاند. في عام 1984، انضمت بروناي دار السلام إلى جيرانها في الجمعية. كمجموعة، يُطلق على هؤلاء المشاركين الأوائل اسم ASEAN6. أصبحت فيتنام عضوًا منذ عام 1995، ولواوس وميانمار منذ عام 1997، وكمبوديا منذ عام 1999. يُشار عادةً إلى هؤلاء الأعضاء الأربعة الجدد باسم مجموعة CLMV.
تم إنشاء مجموعة العمل الدوائي (PPWG) للعمل على التفاصيل في وضع إرشادات التوحيد للإجراءات التقنية والمتطلبات جزئيًا قابلة للتطبيق على الصناعات الدوائية في ASEAN. تم تحديد المجالات التي يمكن توحيدها – الجودة والفعالية والسلامة والبيانات الإدارية.
الوثائق الرئيسية التي نتجت عن عمل PPWG تشمل:
– المتطلبات التقنية المشتركة في ASEAN (ACTR) لتسجيل المنتجات الدوائية (للاستخدام البشري).
– ملف تقني موحد في ASEAN (ACTD) لتسجيل المنتجات الدوائية (للاستخدام البشري).
– إرشادات ASEAN في مجالات مثل التحليل التحليلي ودراسات البيولوجية والتكافؤ والتحقق من العملية والاستقرار.
بينما كانت ASEAN تعمل على تسجيل الدوائي الموحد، يوجد تعاون دولي آخر لتوحيد تسجيل الدوائي يجري في الوقت نفسه يُعرف باسم المؤتمر الدولي لتوحيد المتطلبات التقنية لتسجيل الأدوية للاستخدام البشري (ICH). تأسس في عام 1990 ويعمل على تطوير الإرشادات التقنية لتسجيل منتجات الأدوية لتحقيق توحيد أكبر.
منتجات الأدوية في ASEAN
في أول اجتماع لـ PPWG، تم الاتفاق على مدى الإشراف وتقرر أن تكون المواضيع المحددة للتوحيد مقسمة إلى سلامة وجودة وفعالية وستكون أساساً لاعتماد المنتجات الطبية. إحدى المواضيع الرئيسية لـ PPWG هي فكرة “منتج دوائي في ASEAN”. وهذا يعني أن نفس المتطلبات التنظيمية تنطبق على تسجيل منتج دوائي في جميع الدول الأعضاء في ASEAN. قامت PPWG بتطوير قاموس ASEAN للمصطلحات، و ACTD، و ACTR وإرشاداتها.
يقدم ACTD معلومات حول تنسيق وهيكل الدستور الذي يجب استخدامه بشكل مشترك للتطبيقات في منطقة ASEAN. يجب أن يكون ACTD موجهًا للوثائق التي تم تجميعها لطلب ترخيص تسويق. لا تقدم أي توصيات بشأن المحتوى الفعلي للدستور. يشبه ACTD إلى حد كبير الجزء 2B من ملاحظة مقدمي الطلبات الأوروبية حول العرض والتنسيق للدستور (EU-CTD).
أما ACTR فهو مجموعة من المواد الكتابية تهدف إلى توجيه المتقدمين لإعداد طلب بطريقة تتسق مع توقعات جميع سلطات تنظيم الأدوية في ASEAN. إنها دليل لإعداد ACTD.
يتألف الدليل من أربعة أجزاء:
– الجزء الأول: جدول المحتويات والبيانات الإدارية ومعلومات المنتج
– الجزء الثاني: وثيقة الجودة
– الجزء الثالث: وثيقة غير سريرية
– الجزء الرابع: وثيقة سريرية
يتم تقديم المعلومات أعلاه في هيكل تنظيمي يُعرف باسم مثلث ACTD.
يتم تفصيل الهيكل التنظيمي بمساعدة البيانات الفنية لوثيقة الأمانة التقنية المشتركة في جمعية دول جنوب شرق آسيا (ACTD) التي ذُكرت في جميع أجزاء الملف الخاصة به.
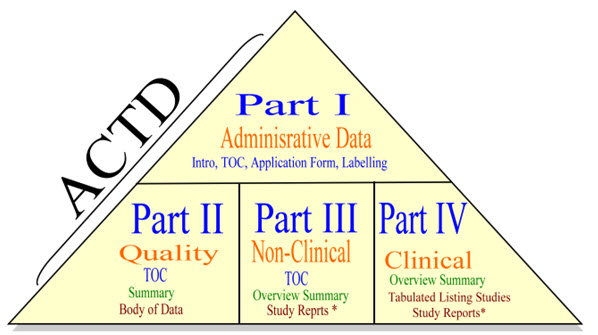
من المقال يمكن استنتاج أن الحصول على ترخيص السوق لتسجيل دواء في أي إقليم يتطلب تنسيقًا معينًا، وأن كل بلد يتبع إرشادًا معينًا بالإضافة إلى لوائحه الخاصة التي وضعتها السلطة التنظيمية للأدوية المعنية. هناك أساساً نوعين من التنسيقات المتاحة في معظم بلدان العالم، واحدة هي ICH-CTD والأخرى هي ACTD. يتم اتباع ICH-CTD في بلدان ICH، بينما يتم اعتماد ACTD عادةً من قبل دول جمعية دول جنوب شرق آسيا (ASEAN). يتألف ICH-CTD من خمسة وحدات، حيث الوحدة 1 تحتوي على معلومات إدارية محددة للبلد، بينما الأربع وحدات الأخرى مخصصة للأدوية وتتضمن تفاصيل علمية وصفية وتعتبر مشتركة بين جميع البلدان التي تتبع هذا التنسيق. الوحدات 2 و 3 و 4 و 5 تحتوي على ملخص للمعلومات حول الجودة والدراسات غير السريرية والسريرية على التوالي. أما ACTD فتتألف من أجزاء بدلاً من وحدات. توجد أربعة أجزاء، تتضمن ملخصًا للجودة وللدراسات غير السريرية والسريرية في الأجزاء II و III و IV على التوالي.